
Valley families struggle to cope with cystic fibrosis

So pretty. So bright. So vivacious. So loved. So threatened.
So it is to be 7-year-old Elizabeth Church.
“I love to play bingo,” the smiling straight-A student says as her older sister Olivia and younger brother Evan join in scrambling for game boards.
The early evening sun slants through a family room window to place the children in a natural spotlight in their North Las Vegas home. Casey and Janice Church laugh, enjoying their children’s enthusiasm.
At first glance, this is a slice of Americana where problems are few, where hard-working parents still parent, where their well-mannered children are all blessed with rosy cheeks and giggles and the high energy of good health.
But appearances, particularly among families where cystic fibrosis is present, are deceiving.
It is true that Elizabeth looks like she stepped off the pages of a clothing catalog, rides bikes, roller blades, goes to ballet lessons, and often yells “Bingo” with the gusto of someone who just won millions. It is also true that she has cystic fibrosis, an inherited disease that causes the body to produce thick, sticky mucus that clogs the lungs and digestive system. The condition spawns hacking coughs, too frequent bowel movements, and recurrent infections that often require hospitalization.
That is why Janice Church, before beginning the nightly bingo game, hands her daughter an inhaler and places what looks much like a life vest over her red-and-white dress. Since morning, Elizabeth has taken about 10 medications.
The vest, hooked up to a power source, begins vibrating, compressing and releasing Elizabeth’s chest wall up to 20 times per second, loosening up mucus that can clog her lungs. It is the second time on this day that she will use the vest for nearly an hour.
IMPROVED TREATMENT
In the 1950s, patients who battled the frequent infections of cystic fibrosis seldom lived past age 5. By the 1980s, regardless of new medications, few made it past their high school graduation.
Today, thanks to new therapies, newborn screening programs that allow treatment to begin sooner, and specialized care centers like the one at University Medical Center, the median age of survival is around 37 years of age. Many patients still die from respiratory failure or cor pulmonale, where there is an increase in blood pressure in the pulmonary artery, the vessel that carries blood from the heart to the lungs. This leads to enlargement and subsequent failure of the right side of the heart.
At the nationally accredited UMC Cystic Fibrosis Center, a team of caregivers helps patients and their families with everything from the latest treatments and proper nutrition to assistance with emotional issues and insurance. If patients don’t have insurance, the team finds programs to pay for medications that can run as high as $10,000 a month.
The team, which includes a social worker and dietitian, sees patients at least every three months .
“We literally wouldn’t be where we are to day without the UMC (cystic fibrosis) team,” Janice Church said. “They go above and beyond.”
When Casey Church, an Air Force tech sergeant, was about to be reassigned in 2009 from Nellis Air Force Base to Minot, N.D., the UMC cystic fibrosis program director, Dr. Craig Nakamura, and program coordinator Tara Brascia wrote a letter to military authorities listing 26 examples of how cystic fibrosis was having an impact on Elizabeth’s life.
Explaining that she showed signs of disease progression and documenting 11 of her daily medications and therapies, the UMC staffers wrote: “In North Dakota, the closest pediatric pulmonologist is 100 miles a way in Bismarck. One hundred miles with a child in respiratory distress is of extreme concern if the child is healthy, but with a child with chronic lung disease we are asking for trouble … moving to North Dakota should NEVER be an option for this family.”
The Air Force pulled its transfer orders. Church, who has served in Iraq and Afghanistan, is grateful to UMC and the military.
“Elizabeth is able to continue to get the care she needs,” he said . “That’s what is important.”
The UMC center takes care of nearly 170 cystic fibrosis patients from Southern Nevada, Utah and Arizona.
When program coordinator Brascia started at the center 20 years ago, only 15 percent of the patients were older than 18.
“Because of new drugs and therapies, 45 percent are now over 18. But it obviously remains a devastating disease. There is so much families must go through every day. And I have seen families where two or three children have it.”
GENETIC MUTATION
Approximately 30,000 people in the United States, and 70,000 people worldwide, have been diagnosed with cystic fibrosis . It is caused by mutations in a gene on chromosome 7, one of the 23 pairs of chromosomes that children inherit from their parents.
Cystic fibrosis occurs most frequently in Caucasians of Northern European descent, although people of other heritages get it as well. Experts say about 12 million Americans, or 1 in every 20 people, is a carrier of the disease, and most don’t know it. Genetic testing is available for couples, but because there are hundreds of specific cystic fibrosis mutations, testing won’t detect everyone who is carrying a defective gene.
During pregnancy, doctors can perform tests so prospective parents can find out more about the chances that their child will have the disease.
To have the disease, a person must inherit two copies of the defective gene, one copy from each parent. When both parents carry the gene, with each pregnancy there is a 25 percent chance that their child will have cystic fibrosis. There is a 50 percent chance that the child will be a carrier like the parents. And there is a 25 percent chance that the baby will not have the gene .
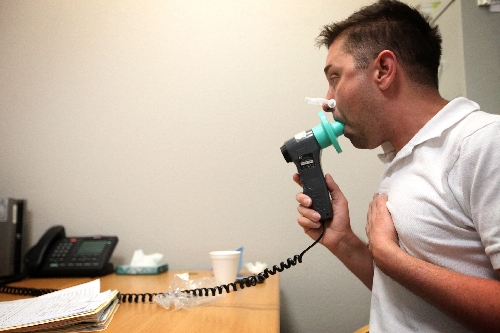
Jamie Thoronton , a Las Vegas banker, was at the UMC center Wednesday for treatment. His older brother died two years ago of the disease at the same age Thoronton is now: 29. His younger brother also has it.
As he was being tested last week for lung function, Thoronton’s coughs left him bent over and gasping for breath.
“This isn’t easy,” he told respiratory therapist Sherry Jackson.
Once he caught his breath, Thoronton said his coughing has long been something he has had to explain.
“In school some students reacted like I have the plague when I cough. And I have to explain that you can’t catch what I’ve got. Most people understand when I say I have a lung disease. My customers at the bank are pretty good about it.”
Brascia often goes to schools to educate teachers, administrators and students about the disease. “The more people learn about it, the better off children with cystic fibrosis are.”
NOT CONTAGIOUS
Cystic fibrosis is not contagious. The cough is simply the body’s defense system against chronic infections. Brascia reminds young patients to keep tissues nearby because it is important to expel the mucus that can lead to infections.
She also lets educators know that students should feel free to leave class until a coughing attack is over.
Because of digestive problems that the disease causes, Brascia also tells educators to allow students with cystic fibrosis to leave the classroom at any time to use the restroom. And to give the students extra time to eat.
It was problems with digestion that indicated to Georgia Cox that her newborn son, Matthew, had a serious problem.
“Everything he ate just went through him,” she recalled.
She was shocked by the diagnosis. “I had never heard of the disease.”
Thick mucus that cystic fibrosis creates also affects the pancreas, an organ that produces proteins called enzymes that flow into the intestine to support the body’s digestive process. The mucus often blocks the path between the pancreas and the intestines, so people with cystic fibrosis have trouble digesting food and getting the vitamins and nutrients they need. Often, the liver is also compromised.
Before digestive therapy — the replacement of enzymes and nutrients — became a regular part of treatment for youngsters , many children with the disease died of malnutrition.
Even with enzyme replacement, vitamin therapy, nutritional drinks and eating high-fat, high-calorie foods, cystic fibrosis patients often have a difficult time getting nourishment. Doctors have to order intravenous nutrient supplementation or feeding tubes through the stomach. Few patients with the disease are overweight.
Elizabeth needs 3,000 calories a day just to stay healthy. That’s double the normal requirement for 7-year-olds.
Georgia Cox still worries that her son, Matthew, now 29 and a practicing attorney in Las Vegas, doesn’t get the nourishment he needs. “He’s too thin,” she said.
Matthew Cox sat in his law office near downtown Las Vegas recently and talked about living with the condition. A former basketball star at Chaparral High School and Antelope Valley College in California, he nearly died two years ago when he had an allergic reaction to an antibiotic for an infection.
Getting up every morning around 5 a.m. for an hourlong breathing treatment before school or work gets old sometimes. So does the breathing treatment at night and the long list of medications he must take. “You really have to schedule your day,” he said.
Like many cystic fibrosis patients, he can give himself IVs full of antibiotics outside the hospital.
“I’ve got my IV pole right here in my office,” said Cox, who has been hospitalized on a few occasions, including during his study at UNLV’s Boyd School of Law.
Cox has noticed that his health has begun to deteriorate over the last couple years, largely because he has stopped vigorous aerobic exercise, a key factor in keeping cystic fibrosis patients as healthy as possible.
“I need to exercise but I start the day so early and often don’t get home till six or seven, so it’s difficult,” he said.
Dr. Nakamura, director of the cystic fibrosis center at UMC, said he thinks the long-term prognosis for Cox is good.
“He’ll make it past the median age (37), I’m sure,” said Nakamura, who is also confident that new drugs are in the pipeline that will greatly expand the life span of people with the disease.
At this point, Cox sees himself making it to 55.
END-OF-LIFE ISSUES
Cystic fibrosis patients routinely talk about end-of-life issues.
T.J. Paxton, 31, sat in the UMC treatment center and recalled how he was on a ventilator for weeks about five years ago. He had expected to die at that point. “I was all right with it. I was at peace.”
To his surprise, he got off the ventilator and ended up getting a lung transplant at UCLA.
“I really did it for my family then,” he said. “But I want to live now.”
In the last few weeks, he has noticed problems with his breathing.
“I can’t understand it,” he told Jackson, the respiratory therapist. “I’ve never had breathing problems before.”
Some people who have severe lung damage caused by cystic fibrosis get a lung transplant. While it helps the patient with breathing because the new lungs are not damaged, the transplant does not prevent or improve any problems that cystic fibrosis may be causing in other parts of the body.
About half of cystic fibrosis patients who receive lung transplants survive at least five years after the procedure. That’s about the same percentage as those who receive a transplant for other reasons.
Jamie Thoronton was going through tests Thursday to see whether he might qualify for a lung transplant. His wife of three years, Autumn, looked on.
“I’m not sure whether it would be the best thing for me or not,” he said.
Before they became romantically involved, Autumn Thoronton learned about the condition and saw how he handled it.
“I’ve always appreciated the way he didn’t let it get in his way, that he didn’t feel sorry for himself. That had a lot to do with the relationship growing.”
The fact that cystic fibrosis causes more than 95 percent of men with the disease to become infertile didn’t make Autumn Thoronton hesitant to marry her husband.
“If children are meant to be, they will be,” she said, explaining that she and her husband are Mormons who find their faith helps them deal with the challenges of cystic fibrosis.
Even before Janice Church gave birth to Elizabeth, she knew how difficult cystic fibrosis could be.
“I had a first cousin who passed with it as a child,” she said.
She broke down in tears when a test done during her pregnancy showed Elizabeth probably would have the disease.
Two weeks after Elizabeth was born, she almost died from an infection.
“The first couple years were really rough,” Church said.
On Wednesday evening, Casey and Janice Church, who read medical publications to stay up on the latest treatment advances in the disease, did what they always do with Elizabeth: integrate her into everything the family does.
“B-3,” Janice Church called out loudly in the bingo game, managing to be heard over Elizabeth’s pulsating vest, which sounds like the “whump whump” of Huey helicopter rotors.
After the game, the mother said Friday, she told the Lord how blessed she was to have such a beautiful family.
“And I did what I do every day: I prayed that He would help scientists find a way to save all kids from cystic fibrosis. I believe my prayers will be answered.”
Contact reporter Paul Harasim at
pharasim@reviewjournal.com or 702-387-2908.









